
This week, we are sharing a report by the MycoLab regarding piglet microbiota and its potential influence of M.hyopneumoniae susceptibility.
Key points
- Early life gut microbiota could be a potential determinant in modulating susceptibility to chronic respiratory diseases such as enzootic pneumonia in pigs.
- Increased abundance of short‐chain fatty acid producing bacteria in piglet gut was associated with decreased M. hyopneumoniae respiratory lesions.
- Understanding the function and composition of a ‘healthy’ pig gut microbiota would aid to successfully implement novel disease control strategies.
Mycoplasma hyopneumoniae is the causative agent of porcine enzootic pneumonia and considered as one of the most important etiologies associated with the chronic respiratory illnesses in swine production. Recent research indicates that a state of compromised gut microbiota could be associated with increased incidence of respiratory infections in animals and humans. Alternatively, early‐life gut microbiota diversity and composition have been identified as predictive biomarkers for respiratory health and disease. Therefore, an exploratory study was conducted to answer whether the early gut microbiome composition of pigs could modulate susceptibility to M. hyopneumoniae infection. The potential association between the presence and abundance of distinct bacterial communities in the piglet gut at different time points, and subsequent susceptibility to develop mycoplasma pneumonia lesions was analyzed using the 16S rRNA fecal microbiome profiling of M. hyopneumoniae infected pigs.
Thirty-two conventional crossbred piglets, from six different litters, were included in this study. Piglets were fed a commercial diet ad libitum and were administered Ceftiofur (5 mg/kg, IM) at three weeks of age. Among them, 30 piglets were experimentally inoculated intra‐tracheally with M. hyopneumoniae strain 2327 (105 CCU/ml) at eight weeks of age. Two piglets remained uninoculated as controls throughout the study. Fecal bacterial community compositions of piglets at three weeks of age, eight weeks of age (prior to M. hyopneumoniae challenge) and twelve weeks of age (four weeks post‐challenge) were targeted by amplifying the V3‐V4 region of 16S rRNA gene. Illumina MiSeq generated sequences were processed using DADA2 workflow and microbiome community analyses were conducted using R software. Lung lesion scores (LS) were recorded as described in European Pharmacopoeia from pig lungs at necropsy.
Although all piglets were challenged with same strain of M. hyopneumoniae, using the same dose intra‐tracheally, piglets from different litters showed varied susceptibility to the pathogen. The LS ranged from 0.3% to 43% with an evident clustering of the scores observed in piglets within litters. For example, piglets from one litter showed the highest mean LS (20%; 95%CI: 30.32–10.4%) when challenged with M. hyopneumoniae, whereas another litter showed the lowest mean LS (2.5%; 95%CI: 4.7 – 0.31%). There were significant differences in the diversity and composition of fecal microbiomes among piglets within litters at different time points (p<0.05). The fecal microbiota of piglets was more dissimilar at week three compared to those from weeks eight and 12 of age.
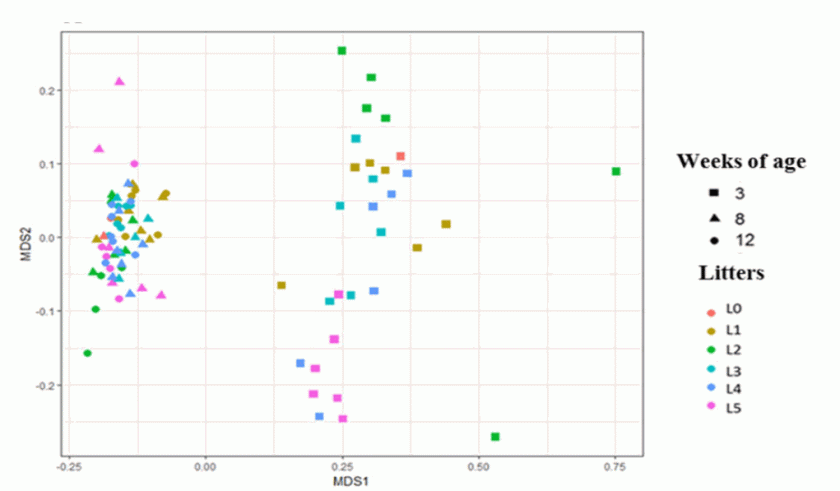
Furthermore, specific groups of bacteria in the gut that might predict the decreased severity of M. hyopneumoniae associated respiratory complications were identified. At three weeks of age, the majority of significantly abundant taxa in piglets who developed low LS belonged to short chain fatty acid (SCFA) producing taxa. For instance, in piglets from the litter with low LS the microbial shift at week three was mainly driven by an increase in abundance of the SCFA bacterial family, Ruminococcaceae. Previously, in rodent models, it has been shown that SCFAs could enter the circulation, modulate bone‐marrow hematopoiesis and thereby could promote regulatory or pro‐inflammatory responses in the lung. Henceforth, our findings on variability of the microbial patterns in early life and its relationship to M. hyopneumoniae exposure highlight prospective opportunities for both nutritional and therapeutic disease control interventions.
Follow up studies are currently underway including information on dam microbiota and cross‐fostering as covariates to explain the observed clustering among the littermates and variation between different litters.