
This is our Friday rubric: every week a new Science Page from the Bob Morrison’s Swine Health Monitoring Project. The previous editions of the science page are available on our website.
In this week’s science page researchers Mariana Kikuti, Carles Vilalta, Juan Sanhueza, Nakarin Pamornchainavakul, Jessica Kevill, My Yang, Igor A. D. Paploski, Tatiana Lenskaia, Nkechi M. Odogwu, Ross Kiehne, Kimberly VanderWaal, Declan Schroeder and Cesar A. Corzo share results of further analysis a 2020 study using whole genome sequencing to assess PRRSv diversity within and across litters.
We have shared preliminary results assessing PRRSV whole-genome viral diversity data over time within the host and within-farm on a previous science page (October 2020). This cohort study was conducted at one naïve farrow-to-wean farm reporting a PRRSV outbreak. All piglets 3–5 days of age (DOA) born to mass-exposed sows through live virus inoculation with the recently introduced wild-type virus two weeks prior were sampled and followed up at 17–19 DOA. Samples from 127 piglets were individually sequenced. We found that percent nucleotide differences within the piglet population was generally small, but higher diversity was found in ORFs 4 and 5a. Here, we share results of further analysis assessing genetic diversity together with piglet mortality.
Piglets that died between the 3–5 and 17–19 DOA samplings had significantly lower median Ct values at baseline than those that survived (14.0 vs. 15.5, p = 0.001) (Figure 1A). The effect of sex and Ct value from the first sampling on the probability of dying is illustrated in Figure 1B. This potentially suggests a complex relationship between sex and viremia, and that these differences found earlier in life might be diluted over time by constant exposure to the virions being shed by this pig population since there was no difference in Ct values or in the mortality rate by sex at 17–19 DOA.

The nucleotide diversity of ORFs 2–7 was calculated with a sliding window of 31 nucleotides and one nucleotide step size and then standardized to the sequence length to obtain the nucleotide diversity of each 31-nucleotide sliding window in relation to the overall genome. Similarly, the same methodology was applied to the translated sequences to estimate regions with increased amino acid diversity using a sliding window of 11 amino acids and one amino acid step size. An arbitrary threshold representing the 90th percentile of the nucleotide/amino acid diversity was set. We observed 10 regions with increased nucleotide diversity (Figure 2A) and eight regions with increased amino acid diversity (Figure 2B). The only ORF5 region in which nucleotide and amino acid diversity were found to be higher than the threshold was between amino acid positions 162–171, which are contained in regions potentially associated with T-cell and B-cell epitopes.
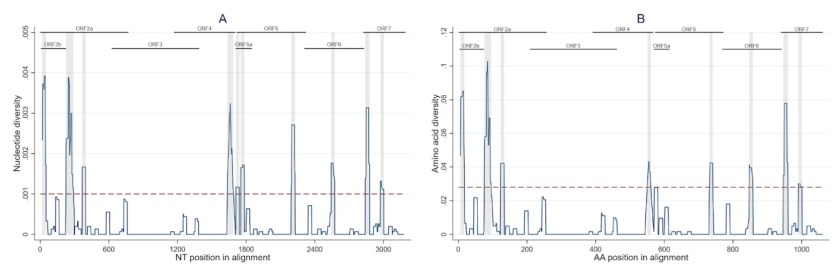
Results from this study highlight the fact the virus evolves over short periods of time within the pig and litter and in specific regions of its genome. More work needs to be conducted to further understand the implications of these changes. The full paper describing all our findings is available at https://www.mdpi.com/1999-4915/15/9/1837